 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings 2026
Congrès & Meetings 2026 Authors
Masson, Jean-Daniel, Taglietti, Valentina, Ruby, François, Ono, Hiroya, Mouri, Nadir, Jorge, Alan, Guillaud, Laurent, Tiret, Laurent, Relaix, Frederic
Lab
Journal
Skeletal Muscle
Abstract
Duchenne muscular dystrophy (DMD) mainly affects young boys with out-of-frame mutations in the DMD gene, leading to dystrophin deficiency. This loss disrupts the assembly of the sarcolemmal dystrophin-associated glycoprotein complex, resulting in membrane fragility and damage during muscle contraction-relaxation cycles. Consequently, patients experience progressive muscle weakness, loss of ambulation and cardiorespiratory failure. Gene therapy represents one of the most promising therapeutic approaches, requiring rigorous preclinical validation of candidate strategies. While several preclinical models of dystrophin deficiency mimic point mutations or exon deletions, no existing rat model accurately replicates DMD gene duplications, which account for approximately 10% of DMD cases. Using CRISPR/Cas9 genome editing, we generated a ~ 125 kbp duplication encompassing exons 10–17 of the Dmd gene in Sprague Dawley rats. To characterise disease progression in these rats, we assessed biochemical, histological and functional biomarkers at 6 and 10 months of age, comparing them to their healthy littermates. We established the R-DMDdup10-17 line. The microstructure of limb, diaphragm and cardiac muscles of R-DMDdup10-17 (DMD) rats exhibited dystrophic changes at 6 and 10 months, including loss of myofibres and fibrosis. These alterations led to a significant body mass reduction, muscle weakness (including diaphragm deficiency) and cardiac electrical defects. Premature lethality was observed between 10 and 13 months. Duplication of the Dmd genomic region encompassing exons 10 to 17 in rats results in dystrophin deficiency, severe striated muscle dystrophy, and premature death. The R-DMDdup10-17 line represents the first reported genetic model of a severe and early lethal duplication variant in the Dmd gene. It provides a critical tool for assessing targeted gene therapies aimed to correct such mutations.
Keywords/Topics
Congenital myopathy; Neuromuscular disorder; Inter-individual data correction; Myonecrosis; MYOM3; hs-cTnT; Plethysmography; ECG; Notched T wave
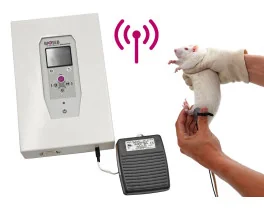
BIOSEB Instruments Used:
Grip strength test (BIO-GS4)
Source :
https://link.springer.com/article/10.1186/s13395-025-00386-2
