 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings 2026
Congrès & Meetings 2026 Authors
J. E. Davies, S. Sarkar, D. C. Rubinsztein.
Lab
Addenbrooke's Hospital, Cambridge Institute for Medical Research, Department of Medical Genetics, Cambridge, UK.
Journal
Human Molecular Genetics
Abstract
Oculopharyngeal muscular dystrophy (OPMD) is a late-onset, progressive disease caused by the abnormal expansion of a polyalanine tract-encoding (GCG)n trinucleotide repeat in the poly-(A) binding protein nuclear 1 (PABPN1) gene. OPMD is generally inherited as an autosomal dominant disorder and the polyalanine expansion mutation is thought to confer a toxic gain-of-function on mutant PABPN1 which forms aggregates within skeletal myocyte nuclei. Here we describe a novel beneficial function of wild-type PABPN1. Wild-type PABPN1 over-expression can reduce mutant PABPN1 toxicity in both cell and mouse models of OPMD. In addition, wild-type PABPN1 provides some protection to cells against pro-apoptotic insults distinct from the OPMD mutation such as staurosporine treatment and Bax expression. Conversely, PABPN1 knockdown (which itself is not toxic) makes cells more susceptible to apoptotic stimuli. The protective effect of wild-type PABPN1 is mediated by its regulation of X-linked inhibitor of apoptosis (XIAP) protein translation. This normal activity of PABPN1 is partially lost for mutant PABPN1; elevated levels of XIAP are seen in mice expressing a wild-type but not a mutant PABPN1 transgene. This raises the possibility that a compromise of the anti-apoptotic function of PABPN1 might contribute to the disease mechanism of OPMD.
BIOSEB Instruments Used
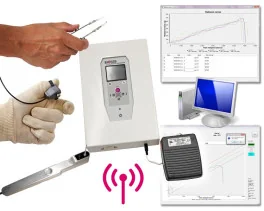
Grip strength test (BIO-GS3)
Keywords/Topics
Domaines de recherche divers
Source :
