 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings
Congrès & Meetings Authors
J Scekic-Zahirovic, M Fischer, GS Lopez, et al
Lab
Journal
Progress in Neurobiology
Abstract
Amyotrophic lateral sclerosis (ALS) arises from the combined degeneration of motor neurons (MN) and corticospinal neurons (CSN). Recent clinical and pathological studies suggest that ALS might start in the motor cortex and spread along the corticofugal axonal projections (including the CSN), either via altered cortical excitability and activity or via prion-like propagation of misfolded proteins. Using mouse genetics, we recently provided the first experimental arguments in favour of the corticofugal hypothesis, but the mechanism of propagation remained an open question. To gain insight into this matter, we tested here the possibility that the toxicity of the corticofugal projection neurons (CFuPN) to their targets could be mediated by their cell autonomous-expression of an ALS causing transgene and possible diffusion of toxic misfolded proteins to their spinal targets. We generated a Crym-CreERT2 mouse line to ablate the SOD1G37R transgene selectively in CFuPN. This was sufficient to fully rescue the CSN and to limit spasticity, but had no effect on the burden of misfolded SOD1 protein in the spinal cord, MN survival, disease onset and progression. The data thus indicate that in ALS corticofugal propagation is likely not mediated by prion-like mechanisms, but could possibly rather rely on cortical hyperexcitability.
BIOSEB Instruments Used
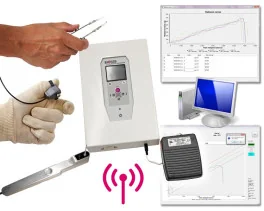
Grip strength test (BIO-GS3)
Source :
