 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings 2026
Congrès & Meetings 2026 Authors
A. Buj-Bello, V. Laugel, N. Messaddeq, H. Zahreddine, J. Laporte et al.
Lab
Institut de Génétique et de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique/Institut National de la Santé et de la Recherche Médicale/Université Louis Pasteur Strasbourg, Illkirch, France.
Journal
PNAS, Proceedings of the National Academy of Sciences of the United States of America
Abstract
Myotubularin is a ubiquitously expressed phosphatase that acts on phosphatidylinositol 3-monophosphate [PI(3)P], a lipid implicated in intracellular vesicle trafficking and autophagy. It is encoded by the MTM1 gene, which is mutated in X-linked myotubular myopathy (XLMTM), a muscular disorder characterized by generalized hypotonia and muscle weakness at birth leading to early death of most affected males. The disease was proposed to result from an arrest in myogenesis, as the skeletal muscle from patients contains hypotrophic fibers with centrally located nuclei that resemble fetal myotubes. To understand the physiopathological mechanism of XLMTM, we have generated mice lacking myotubularin by homologous recombination. These mice are viable, but their lifespan is severely reduced. They develop a generalized and progressive myopathy starting at around 4 weeks of age, with amyotrophy and accumulation of central nuclei in skeletal muscle fibers leading to death at 6–14 weeks. Contrary to expectations, we show that muscle differentiation in knockout mice occurs normally. We provide evidence that fibers with centralized myonuclei originate mainly from a structural maintenance defect affecting myotubularin-deficient muscle rather than a regenerative process. In addition, we demonstrate, through a conditional gene-targeting approach, that skeletal muscle is the primary target of murine XLMTM pathology. These mutant mice represent animal models for the human disease and will be a valuable tool for understanding the physiological role of myotubularin.
BIOSEB Instruments Used
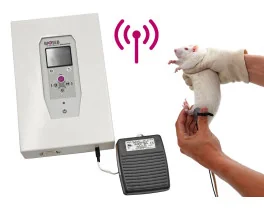
Grip strength test (BIO-GS3)
Keywords/Topics
Myopathie; Système musculaire
Source :
