 Douleur - Allodynie/Hyperalgésie Thermique
Douleur - Allodynie/Hyperalgésie Thermique Douleur - Spontanée - Déficit de Posture
Douleur - Spontanée - Déficit de Posture Douleur - Allodynie/Hyperalgésie Mécanique
Douleur - Allodynie/Hyperalgésie Mécanique Apprentissage/Mémoire - Attention - Addiction
Apprentissage/Mémoire - Attention - Addiction Physiologie & Recherche Respiratoire
Physiologie & Recherche Respiratoire





























 Douleur
Douleur Système Nerveux Central (SNC)
Système Nerveux Central (SNC)  Neurodégénérescence
Neurodégénérescence Système sensoriel
Système sensoriel Système moteur
Système moteur Troubles de l'humeur
Troubles de l'humeur Autres pathologies
Autres pathologies Système musculaire
Système musculaire Articulations
Articulations Métabolisme
Métabolisme Thématiques transversales
Thématiques transversales Congrès & Meetings 2026
Congrès & Meetings 2026 Authors
A. Vignaud, A. Ferry, A. Huguet, M. Baraibar, C. Trollet et al.
Lab
Université Pierre et Marie Curie, Paris, France.
Journal
Neuromuscular Disorders
Abstract
Myotonic dystrophy type 1 (DM1) is a neuromuscular disease caused by the expansion of a CTG repeat in the DMPK gene and characterised by progressive skeletalmuscleweakness and wasting. To investigate the effects of the CTGexpansion on the physiological function of the skeletalmuscles, we have used a transgenicmouse model carrying the human DM1 region with 550 expanded CTG repeats. Maximal force is reduced in the skeletalmuscles of 10-month-old but not in 3-month-old DM1 mice when compared to age-matched non-transgenic littermates. The progressive weakness observed in the DM1 mice is directly related to the reduced muscle mass and muscle fibre size. A significant increase in trypsin-like proteasome activity and Fbxo32 expression is also measured in the DM1 muscles indicating that an atrophic process mediated by the ubiquitin–proteasomepathway may contribute to the progressive muscle wasting and weakness in the DM1 mice.
BIOSEB Instruments Used
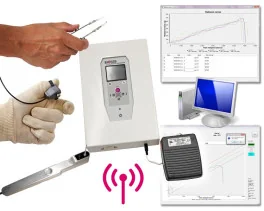
Grip strength test (BIO-GS3)
Keywords/Topics
Phénotypage; Thématiques transversales
Source :
http://www.sciencedirect.com/science/article/pii/S0960896610001112
