 Pain - Thermal Allodynia / Hyperalgesia
Pain - Thermal Allodynia / Hyperalgesia Pain - Spontaneous Pain - Postural Deficit
Pain - Spontaneous Pain - Postural Deficit Pain - Mechanical Allodynia / Hyperalgesia
Pain - Mechanical Allodynia / Hyperalgesia Learning/Memory - Attention - Addiction
Learning/Memory - Attention - Addiction Physiology & Respiratory Research
Physiology & Respiratory Research

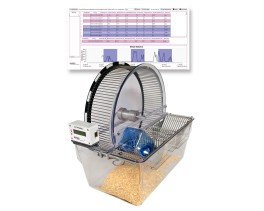









![Dynamic Weight Bearing 2.0 – Postural Module [Add-on]](https://bioseb.com/733-home_default/dynamic-weight-bearing-20-add-on-postural-module.jpg)



















 Pain
Pain Central Nervous System (CNS)
Central Nervous System (CNS) Neurodegeneration
Neurodegeneration Sensory system
Sensory system Motor control
Motor control Mood Disorders
Mood Disorders Other disorders
Other disorders Muscular system
Muscular system Joints
Joints Metabolism
Metabolism Cross-disciplinary subjects
Cross-disciplinary subjects CONFERENCES & MEETINGS 2026
CONFERENCES & MEETINGS 2026 Authors
K Kim, NN Jha, Z Feng, MR Faleiro, CA Chiriboga et al
Lab
Columbia University Medical Center, New York, New York, USA
Journal
Journal of Clinical Investigation
Abstract
Paucity of the survival motor neuron (SMN) protein triggers the oft-fatal infantile-onset motor neuron disorder, spinal muscular atrophy (SMA). Augmenting the protein is one means of treating SMA and recently led to FDA approval of an intrathecally delivered SMN-enhancing oligonucleotide currently in use. Notwithstanding the advent of this and other therapies for SMA, it is unclear whether the paralysis associated with the disease derives solely from dysfunctional motor neurons that may be efficiently targeted by restricted delivery of SMN-enhancing agents to the nervous system, or stems from broader defects of the motor unit, arguing for systemic SMN repletion. We investigated the disease-contributing effects of low SMN in one relevant peripheral organ — skeletal muscle — by selectively depleting the protein in only this tissue. We found that muscle deprived of SMN was profoundly damaged. Although a disease phenotype was not immediately obvious, persistent low levels of the protein eventually resulted in muscle fiber defects, neuromuscular junction abnormalities, compromised motor performance, and premature death. Importantly, restoring SMN after the onset of muscle pathology reversed disease. Our results provide the most compelling evidence yet for a direct contributing role of muscle in SMA and argue that an optimal therapy for the disease must be designed to treat this aspect of the dysfunctional motor unit.
BIOSEB Instruments Used
Grip strength test (BIO-GS3)
Keywords/Topics
Muscular atrophy; Muscular system
Source :
