 Pain - Thermal Allodynia / Hyperalgesia
Pain - Thermal Allodynia / Hyperalgesia Pain - Spontaneous Pain - Postural Deficit
Pain - Spontaneous Pain - Postural Deficit Pain - Mechanical Allodynia / Hyperalgesia
Pain - Mechanical Allodynia / Hyperalgesia Learning/Memory - Attention - Addiction
Learning/Memory - Attention - Addiction Physiology & Respiratory Research
Physiology & Respiratory Research










![Dynamic Weight Bearing 2.0 – Postural Module [Add-on]](https://bioseb.com/733-home_default/dynamic-weight-bearing-20-add-on-postural-module.jpg)



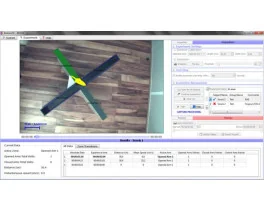
















 Pain
Pain Central Nervous System (CNS)
Central Nervous System (CNS) Neurodegeneration
Neurodegeneration Sensory system
Sensory system Motor control
Motor control Mood Disorders
Mood Disorders Other disorders
Other disorders Muscular system
Muscular system Joints
Joints Metabolism
Metabolism Cross-disciplinary subjects
Cross-disciplinary subjects CONFERENCES & MEETINGS 2026
CONFERENCES & MEETINGS 2026 Authors
Taglietti, V., Kefi, K., Bronisz-Budzy_ska, I. et al.
Lab
Univ Paris-Est Creteil, INSERM, Creteil, France
Journal
Acta Neuropathologica Communications
Abstract
Duchenne muscular dystrophy (DMD) is a fatal muscle-wasting disorder caused by mutations in the Dystrophin gene and for which there is currently no cure. To bridge the gap between preclinical and therapeutic evaluation studies, we have generated a rat model for DMD that carries an exon 52 deletion (R-DMDdel52) causing a complete lack of dystrophin protein. Here we show that R-DMDdel52 animals recapitulated human DMD pathophysiological trajectory more faithfully than the mdx mouse model. We report that R-DMDdel52 rats displayed progressive and severe skeletal muscle loss associated with fibrotic deposition, fat infiltration and fibre type switch. Early fibrosis was also apparent in the cardiac muscle. These histological modifications led to severe muscle, respiratory and cardiac functional impairments leading to premature death around 1 year. Moreover, DMD muscle exhibited systemic inflammation with a mixed M1/M2 phenotype. A comparative single cell RNAseq analysis of the diaphragm muscle was performed, revealing cellular populations alteration and molecular modifications in all muscle cell types. We show that DMD fibroadipogenic progenitors produced elevated levels of cartilage oligomeric matrix protein, a glycoprotein responsible for modulating homeostasis of extracellular matrix, and whose increased concentration correlated with muscle fibrosis both in R-DMDdel52 rats and human patients. Fibrosis is a component of tissue remodelling impacting the whole musculature of DMD patients, at the tissue level but most importantly at the functional level. We therefore propose that this specific biomarker can optimize the prognostic monitoring of functional improvement of patients included in clinical trials.
BIOSEB Instruments Used
Grip strength test (BIO-GS3)
Keywords/Topics
Duchenne muscular dystrophy
Source :
