 Pain - Thermal Allodynia / Hyperalgesia
Pain - Thermal Allodynia / Hyperalgesia Pain - Spontaneous Pain - Postural Deficit
Pain - Spontaneous Pain - Postural Deficit Pain - Mechanical Allodynia / Hyperalgesia
Pain - Mechanical Allodynia / Hyperalgesia Learning/Memory - Attention - Addiction
Learning/Memory - Attention - Addiction Physiology & Respiratory Research
Physiology & Respiratory Research

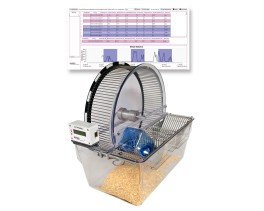









![Dynamic Weight Bearing 2.0 – Postural Module [Add-on]](https://bioseb.com/733-home_default/dynamic-weight-bearing-20-add-on-postural-module.jpg)



















 Pain
Pain Central Nervous System (CNS)
Central Nervous System (CNS) Neurodegeneration
Neurodegeneration Sensory system
Sensory system Motor control
Motor control Mood Disorders
Mood Disorders Other disorders
Other disorders Muscular system
Muscular system Joints
Joints Metabolism
Metabolism Cross-disciplinary subjects
Cross-disciplinary subjects CONFERENCES & MEETINGS 2026
CONFERENCES & MEETINGS 2026 Authors
Emmanuele V, Kubota A, Garcia-Diaz B, Garone C, Akman HO, Sánchez-Gutiérrez D, Escudero LM, Kariya S, Homma S, Tanji K, Quinzii CM, Hirano M
Lab
Istituto di Ricovero e Cura a Carattere Scientifico G. Gaslini, University of Genoa, Genoa 16100, Italy
Journal
Hum Mol Genet.
Abstract
A member of the four-and-a-half-LIM (FHL) domain protein family, FHL1, is highly expressed in human adult skeletal and cardiac muscle. Mutations in FHL1 have been associated with diverse X-linked muscle diseases: scapuloperoneal (SP) myopathy, reducing body myopathy, X-linked myopathy with postural muscle atrophy, rigid spine syndrome (RSS) and Emery-Dreifuss muscular dystrophy. In 2008, we identified a missense mutation in the second LIM domain of FHL1 (c.365 G>C, p.W122S) in a family with SP myopathy. We generated a knock-in mouse model harboring the c.365 G>C Fhl1 mutation and investigated the effects of this mutation at three time points (3-5 months, 7-10 months and 18-20 months) in hemizygous male and heterozygous female mice. Survival was comparable in mutant and wild-type animals. We observed decreased forelimb strength and exercise capacity in adult hemizygous male mice starting from 7 to 10 months of age. Western blot analysis showed absence of Fhl1 in muscle at later stages. Thus, adult hemizygous male, but not heterozygous female, mice showed a slowly progressive phenotype similar to human patients with late-onset muscle weakness. In contrast to SP myopathy patients with the FHL1 W122S mutation, mutant mice did not manifest cytoplasmic inclusions (reducing bodies) in muscle. Because muscle weakness was evident prior to loss of Fhl1 protein and without reducing bodies, our findings indicate that loss of function is responsible for the myopathy in the Fhl1 W122S knock-in mice.
BIOSEB Instruments Used
Grip strength test (BIO-GS3)
Keywords/Topics
Myopathy; Muscular system
Source :
