 Pain - Thermal Allodynia / Hyperalgesia
Pain - Thermal Allodynia / Hyperalgesia Pain - Spontaneous Pain - Postural Deficit
Pain - Spontaneous Pain - Postural Deficit Pain - Mechanical Allodynia / Hyperalgesia
Pain - Mechanical Allodynia / Hyperalgesia Learning/Memory - Attention - Addiction
Learning/Memory - Attention - Addiction Physiology & Respiratory Research
Physiology & Respiratory Research

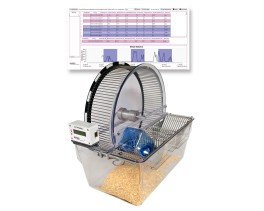









![Dynamic Weight Bearing 2.0 – Postural Module [Add-on]](https://bioseb.com/733-home_default/dynamic-weight-bearing-20-add-on-postural-module.jpg)



















 Pain
Pain Central Nervous System (CNS)
Central Nervous System (CNS) Neurodegeneration
Neurodegeneration Sensory system
Sensory system Motor control
Motor control Mood Disorders
Mood Disorders Other disorders
Other disorders Muscular system
Muscular system Joints
Joints Metabolism
Metabolism Cross-disciplinary subjects
Cross-disciplinary subjects CONFERENCES & MEETINGS 2026
CONFERENCES & MEETINGS 2026 Authors
Bouhy Delphinea, Geuens Thomasa, De Winter Vickya, De Almeida Souza Leonardoa, Katona Istvanb, Weis Joachimb, Hochepied Tinoc, Goossens Stevend, Haigh Jody J.d, Janssens Sophiea, Timmerman Vincenta
Lab
Department of Internal medicine, Gent University, Belgium
Journal
Journal of Neuromuscular Diseases
Abstract
Charcot-Marie-Tooth (CMT) and associated neuropathies, the most common inherited diseases of the peripheral nervous system, remain so far incurable. Three existing murine models of Charcot-Marie-Tooth type 2F (CMT2F) and/or distal hereditary motor neuropathy type IIb (dHMNIIb), caused by mutations in the small heat shock protein B1 gene (HSPB1/HSP27), partially recapitulate the hallmarks of peripheral neuropathy. Because these models overexpress the HSPB1 mutant proteins they differ from the patients’ situation. Objective: To overcome the possible bias induced by overexpression, we generated and characterized a transgenic model in which the wild type or mutant HSPB1 protein was expressed at a moderate, more physiologically relevant level. Methods: We generated a new transgenic mouse model in which a human wild type (hHSPB1WT) or mutant (hHSPB1R127W; hHSPB1P182L) HSPB1 transgene was integrated in the mouse ROSA26 locus. The motor and sensory functions of the mice was assessed at 3, 6, 9, 12 and 18 month. Results: However, the mice expressing the mutant hHSPB1 do not develop motor or sensory deficits and do not show any sign of axonal degeneration, even at late age. Quantitative PCR analyses reveal contrasting tissue-specific expression pattern for the endogenous mouse and exogenous human HSPB1 and show that the ratio of human HSPB1 to the endogenous mouse HspB1 is lower in the sciatic nerve and spinal cord compared to the brain. Conclusion: These results suggest that expressing the transgene at a physiological level using the ROSA26 locus may not be sufficient to model inherited peripheral neuropathies caused by mutation in HSPB1.
BIOSEB Instruments Used
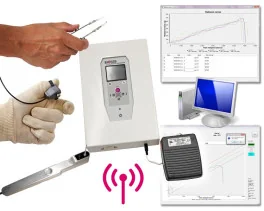
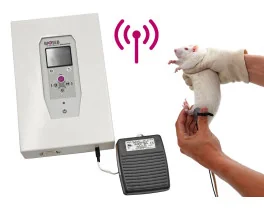
Grip strength test (BIO-GS3)
Keywords/Topics
Miscellaneous research domains
Source :
